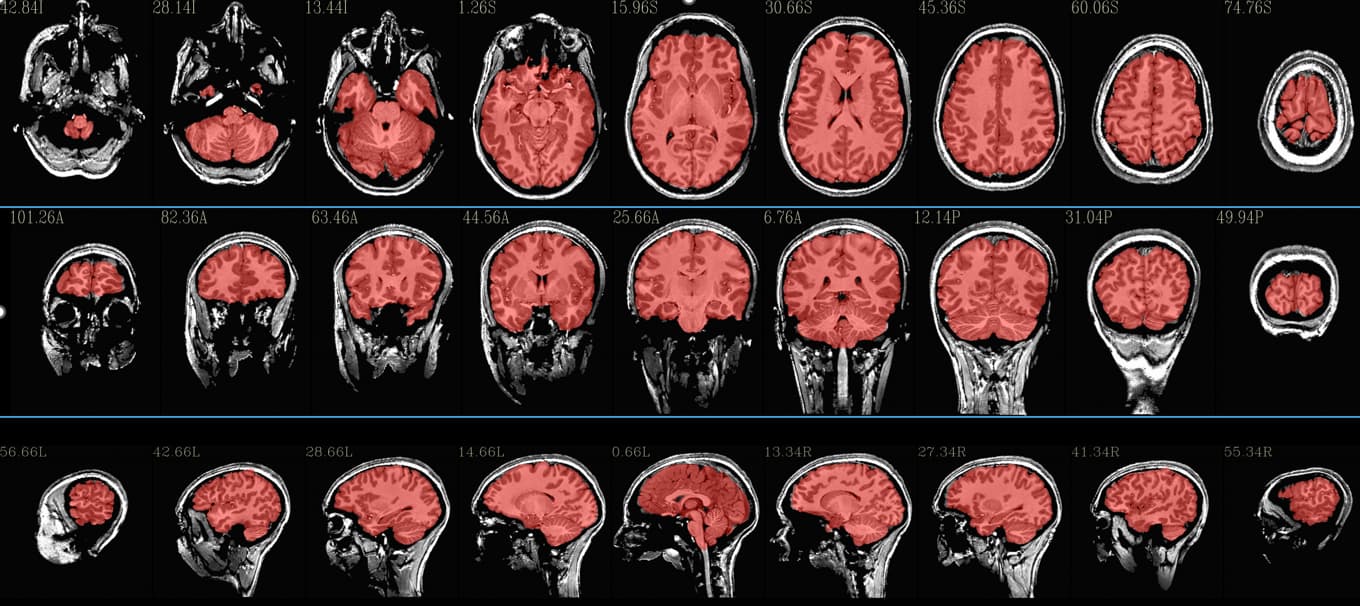
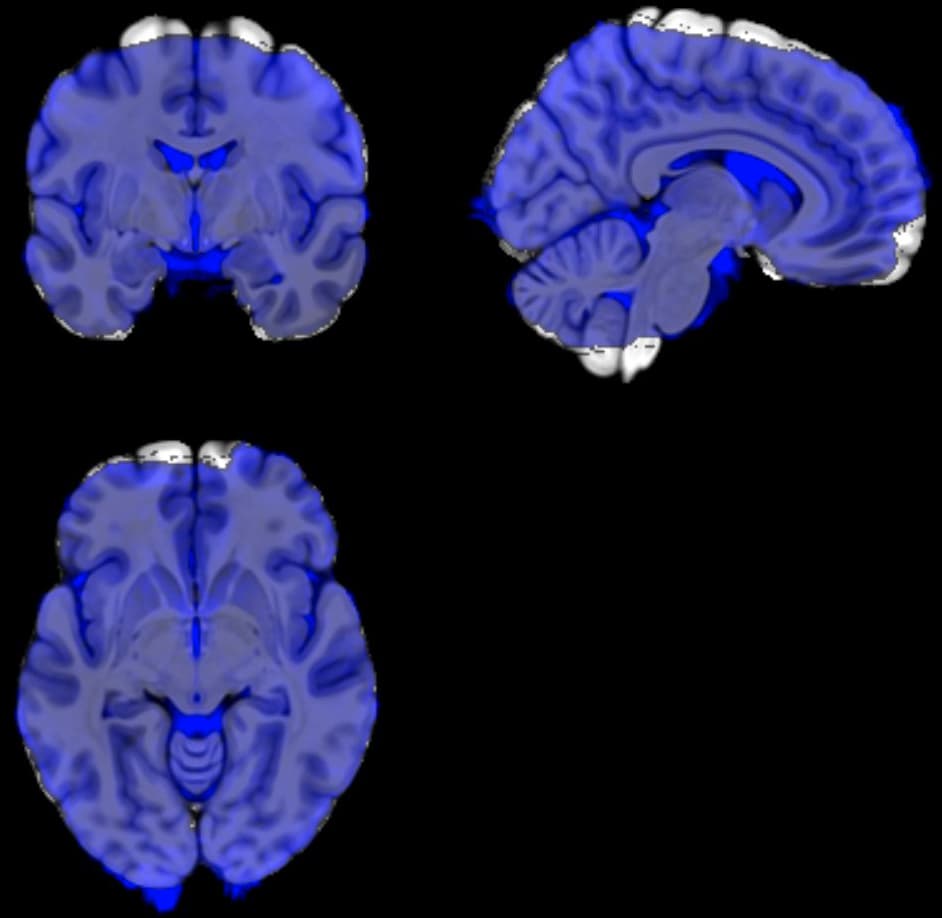
What do the QC images created by @SSwarper look like? Do they look good?
Note that normally the subject T1w anatomical is input into @SSwarper, and that that is aligned to a standard space template---that is something with enough spatial detail to have a good chance of good alignment to template space.
The subject EPI is typically much poorer resolution and contrast, and is not directly aligned to standard space. Instead, one would align the EPI to the anatomical (say, an affine alignment with align_epi_anat.py); align the anatomical to the template (nonlinear alignment with @SSwarper), and then concatenate the warps before applying it to the EPI.
In most FMRI processing, there is the additional alignment step of motion "correction", which is usually rigid body alignment across time. That, too, would be concatenated with the other warps, before applying to the EPI, to minimize blurring from multiple regriddings.
This full barrage of alignments, as well as their proper concatenation, is one of the many convenient things that afni_proc.py will generally take care of for you.
It's not clear to me what the inputs of these various things are. Did you use:
@SSwarper to align the anatomical to template
align_epi_anat.py to align the EPI to the anatomical
(which is basically the standard way of going)? From the two good alignments you show, that appears to be the case.
If so, you need to concatenate all alignment/warp pieces: EPI->anat affine, anat->template affine, anat->template nonlinear warp. From your codes here, it looks like these various alignments that are not good have not done so. Also, note that often with align_epi_anat.py, we align the anat->EPI, so that matrix has to be inverted. But that question depends in detail on the transforms you made.
So, if you could copy+paste your commands that you ran here (both to generate the alignments and to try to apply them), that would help clarify things.
But again, this kind of task would be done most easily using afni_proc.py, where you can even just specify alignment blocks and no regression even, if you don't want (but including that would get you the QC HTML).
@SSwarper produces both an affine transformation and a nonlinear warp - two different files. With EPI to anatomical alignment transformations and motion correction transformations too, all the transformations are chained together into a single transformation. afni_proc.py usually takes care of this complicated procedure - getting the order and direction of each right.
Also I don't think MNI+tlrc is a template we distribute, so it's not clear whether that dataset was used for the alignment in @SSwarper. Post your actual command to do the alignment, maybe sticking to the MNI152_2009_template_SSW.nii.gz as your template base for SSW and 3dNwarpApply. Your MNI+tlrc might be just fine; I'm not familiar with that particular dataset, and there are lots of MNI templates available that can yield different results.
I will clarify things to make it easier for you to see where I am coming from:
I did some resting-state functional connectivity analyses in native EPI space space and now I want to do a group comparison, hence I want to normalize the corrZ maps that I got. That implies that the data have already been preprocessed and analyzed on the 1st level.
What I did so far was using SSwarper on the anatomical image to normalize it to MNI
epi_al_mat.aff12.1D, which should be the EPI-> affine matrix as a specified so in the command. Therefore, an inversion of this matrix is not necessary, I think?
If I understood correctly, I have to somehow concatenate the two matrices epi_al_mat.aff12.1D and anatQQ.sswarp_anat2mni.aff12.1D into one file and use it together with anatQQ.sswarp_anat2mni_WARP.nii in 3dNwarpApply?
My question would be how to concatenate the 2 matrices properly and input it correctly into the 3dNwarpApply command?
Finally, I understand that afni_proc is for proper fMRI preprocessing, which I don't have to use, therefore I was wondering whether it makes sense to use it?
This example includes both the motion correction (volume registration) transformations. In your case, you may decide to leave that out. Still, you would have to be careful to use the same registration base index as the alignment base (anat to EPI). The affine transformations that are together in the chain can be concatenated with cat_matvec. Note that in this example code with the anat to EPI (not EPI to anat) alignment affine transformation, you would invert that for alignment of the EPI to anat instead (that's what the -I ("eye") is for). If using the EPI to anatomical affine, then you would NOT invert that particular affine transformation.
Be careful with the naming of the base template. Your last snippet did not include the full filename.
If you are viewing the transformed volreg base, there should be no difference between them, as the volreg base is not affected by the volreg transformation.
Out of curiosity, why are you not using afni_proc.py for this? Is it for some part of registration that afni_proc.py does not do? Perhaps you could just use afni_proc.py for the 'align tlrc volreg' blocks, at the least, since that seems to be what you are putting together by hand.
@rickr: I assume that afni_proc works with 4D images (time series only) and since I only have on 3D epi image it doesn't work and throws the following error:
3dcopy /data/LC2016/anat+orig 2016.results/anat
++ 3dcopy: AFNI version=AFNI_22.1.08 (May 3 2022) [64-bit]
3dTcat -prefix 2016.results/pb00.2016.r01.tcat /data/2016/epi+orig[0..$]
++ 3dTcat: AFNI version=AFNI_22.1.08 (May 3 2022) [64-bit]
++ elapsed time = 0.4 s
set tr_counts = ( 1 )
cd 2016.results
3dbucket -prefix vr_base pb00.2016.r01.tcat+orig[2]
++ 3dbucket: AFNI version=AFNI_22.1.08 (May 3 2022) [64-bit]
** ERROR: selector index 2 is out of range 0..0
can't decipher index codes from pb00.2016.r01.tcat+orig[2]
I used the following commands to create the script:
Ah, yes, you are probably right here. The output from the align_epi_anat.py is inverted for the epi2anat output, so you wouldn't need to invert it again. With just a single volume in the EPI, you wouldn't be carrying the motion correction with that.
The problem is that the default volume for EPI alignment is the [2]th one, so you need to specify something different. Here, add the opt -volreg_align_to first to your command.
I think you will also need to add this, to turn off a variance check: -find_var_line_blocks NONE. If you aren't seeing that message already, I wonder if your AFNI version is a bit old?
I would also add this, because radial correlation calcs will whine if there is only 1 volume: -radial_correlate_blocks NONE
Finally, I had added this in my test case, but since you aren't really checking for outliers, it might not matter: -outlier_polort 0
You should also specify whether your anatomical has its skull on or not, for alignment: -anat_has_skull no
or -anat_has_skull yes.
That would all lead to this command for your data, with YES_OR_NO needing to be replaced with "yes" or "no":
Also, just to be sure, do you also have a series of EPI volumes that you will be including in this processing/alignment? Because if so, it is worth noting that motion correction is also an alignment process, and therefore should be concatenated in the giant set of alignment transforms before application. And again, this is something that afni_proc.py would handle well, automatically and by design.
thanks again for your responses and help regarding the normalization!
@ptaylor: there will be no time series to normalize because the 1st level analysis (resting-state) has been done in native space and only the 3D Zcorrelation maps (from a seed-based FC analysis) will be normalized for group comparison, hence movement should not be an issue. However, that begs the question of how to normalize a Zcorr map with afni_proc given that it looks necessarily very different from a standard EPI?
In order to include @SSwarper, I'd modify the nice command you created by the following:
If your 1st level analysis is already done, I guess that means you have already done motion correction and the realignment that includes? Note that motion correction itself is an alignment process, one which regrids the EPI volumes. Doing so inherently blurs the data, and then later aligning the data to standard space will blur it again, which is something that should be avoided, if possible. And it is possible, because afni_proc.py would combine all of the following alignments---motion correction, EPI-anatomical and anatomical-template---into a single transform.
I'm curious: how did you perform the first level analysis? Is there a reason to not let afni_proc.py do that, so all the processing features like alignment concatenation---as well as others, and particularly the useful QC checks automatically included in afni_proc.py---can be done for you?
alright, I am happy to go into more detail to give a clearer picture of the situation: overall, I analyse resting-state data in terms of seed-to-whole-brain functional connectivity.
To this end, the data (which are multi echo data) have been preprocessed with multi-echo independent component analysis (ME-ICA; https://pubmed.ncbi.nlm.nih.gov/22209809/ ) with the aim of separating BOLD from non-BOLD signal.
This toolbox actually uses several afni functions, such as 3dretroicor for respiratory and pulse data, 3dTShift+3dvolreg for estimating motion parameters, 3dSkullStip+3dAllineate for anatomical coregistration, where motion correction and anatomical coreg were done in one step using 3dAllineate; 3dBlurInMask is then used for 5mm FWHM smoothing and a high pass filter of 0.02Hz is finally applied before the operations regarding echo combination begin, which include denoising and the ME-ICA component sorting method.
The output file, which is 4D nifti of the preprocessed 4D time series is then subjected to the FC analysis including time series extraction from the seed region with 3dmaskave
This includes an example of a full afni_proc.py script (with @SSwarper run beforehand) for using Kundu et al's MEICA program (sidenote: that was developed down the hallway from the AFNI developers, and has been a part of the AFNI toolbox since about the time it was developed).
You can also implement the optimal combination (OC) echo combination mechanism (Posse et al., 1999), again within afni_proc.py---it's easy to swap those out, and see which you prefer. There are further examples in the AP help.
There is also the much more modern tedana group version of MEICA (Du Pre et al., 2021), which is includable within afni_proc.py.
I think this would greatly simplify and streamline your processing. You can choose what primary parameters you want (blur radius, final voxel size, censoring thresholds, etc.), while letting afni_proc.py doing the more burdensome concatenation/etc. tasks under the hood.
Did you want to do a surface-based analysis? That example you picked is for that, but from previous discussions, it didn't sound like that was what you were headed towards.
What properties do you want for your analysis? For example:
volumetric analysis, or surface-based?
if volumetric, going to standard space, or leaving in subject space?
what form of combining echoes: Kundu et al. MEICA, DuPre et al. MEICA, or Posse et al. OC?
voxelwise analysis, or ROI-based (determines if you want to blur the data or not, respectively)?
Answering those will determine which demo script would be the best starting point. And, because it's afni_proc.py, you could easily switch between those, too, relatively conveniently.
The
National Institute of Mental Health (NIMH) is part of the National Institutes of
Health (NIH), a component of the U.S. Department of Health and Human
Services.