example 13 only caught my eyes, because of the preprocessing steps. As I wrote above, I would like to do a seed-to-whole-brain functional connectivtiy analysis (ROI-based) in native space (1st level) using Kundu et al. MEICA (but also open to DuPre et al. MEICA at a later point) and then normalize the resulting Zcorr maps to do 2nd level analyses.
In this regard, I would be really interested to learn how you normalize Zcorr maps using AP.
There is no need to split "1st level" and "2nd level" things like that. I would think of just "single subject" processing, going to include: volume registration (motion correction), EPI-anatomical alignment, anatomical-template alignment, (possibly blurring and/or estimating masks) and regression modeling. That is basically the set of steps you would specify in the AP blocks. Then you can run 3dNetCorr to calculate whole-brain, seedbased connectivity maps when the "seed" is an ROI-average time series.
Splitting out the step of sending Zscore maps only to standard space (what is sometimes called "normalization" as a shorthand for "spatial normalization", though "normalization" can also be used to refer to "brightness uniformizing") does not seem necessary. Do you need the regression maps only in subject space, while the Z-score/correlation maps are in standard space?
It sounds like the do_23_ap_me_bt.tcsh and do_26_ap_me_bT.tcsh scripts in the APMULTI demo are the best starting point for you. The only difference between these is whether Kundu et al or Du Pre et al MEICA is used, respectively. The only adaptation these might need to your data is that these had a blip up/down correction for phase distortion in them, which you haven't mentioned, so that can be excised by just leaving out that step.
Additionally, you haven't mentioned FreeSurfer, so I removed the use of those follower datasets here (for later ROI-based analysis) as well as tissue-based regressors. Therefore, how about starting with this, for the Kundu et al. MEICA (NB: everywhere NAME_* appears, replace that with sometim appropriate for your data, whether it is a filename, label or numerical value):
set subj = NAME_A
set dir_ssw = NAME_B
set sdir_epi = NAME_C
set dsets_epi_me = ( ${sdir_epi}/${subj}_${ses}_task-rest_*_echo-?_bold.nii* )
set me_times = ( NAME_D NAME_E .... ) # add in values here for echo times
set anat_cp = ${sdir_ssw}/anatSS.${subj}.nii
set anat_skull = ${sdir_ssw}/anatU.${subj}.nii
set dsets_NL_warp = ( ${sdir_ssw}/anatQQ.${subj}.nii \
${sdir_ssw}/anatQQ.${subj}.aff12.1D \
${sdir_ssw}/anatQQ.${subj}_WARP.nii )
# control variables --- SELECT APPROPRIATE VALUES FOR ALL,
set nt_rm = NAME_F # how many initial time points to remove
set blur_size = NAME_G # if using, keep small, since ME and NL warp
set final_dxyz = NAME_H # final output resolution;
set cen_motion = 0.2 # motion censor limit, adjust as desired
set cen_outliers = 0.05 # outlier fraction limit, adjust as desired
afni_proc.py \
-subj_id ${subj} \
-blocks despike tshift align tlrc volreg mask combine blur scale \
regress \
-radial_correlate_blocks tcat volreg regress \
-copy_anat ${anat_cp} \
-anat_has_skull no \
-anat_follower anat_w_skull anat ${anat_skull} \
-dsets_me_run ${dsets_epi_me} \
-echo_times ${me_times} \
-combine_method OC_tedort \
-combine_tedort_reject_midk no \
-tcat_remove_first_trs ${nt_rm} \
-tshift_interp -wsinc9 \
-align_opts_aea -cost lpc+ZZ -giant_move -check_flip \
-tlrc_base ${template} \
-tlrc_NL_warp \
-tlrc_NL_warped_dsets ${dsets_NL_warp} \
-volreg_align_to MIN_OUTLIER \
-volreg_align_e2a \
-volreg_tlrc_warp \
-volreg_warp_final_interp wsinc5 \
-volreg_warp_dxyz ${final_dxyz} \
-volreg_compute_tsnr yes \
-blur_size ${blur_size} \
-mask_epi_anat yes \
-regress_motion_per_run \
-regress_censor_motion ${cen_motion} \
-regress_censor_outliers ${cen_outliers} \
-regress_apply_mot_types demean deriv \
-regress_est_blur_epits \
-regress_est_blur_errts \
-html_review_style pythonic
thank you for the script! I had a careful look at it and your comments and have a few points I would like to clarify. I assume that this script essentially preprocesses the RS data, does ME-ICA and then spatially normalizes them.
One important part of my analysis that I have not strongly pointed out are the seeds: they are individually segmented masks based on a machine learning algorithm that has been applied to a 0.4x0.4x0.4 mm brain stem image. I then transformed the seed masks into EPI space to extract the average time series.
Now, from my understanding, it is better to work in native EPI space as the data are not affected by the spatial normalization procedure, i.e. interpolation confounds. However, it seems that with the way AP works here, the final output is in MNI space and I would have to normalize the seed masks and then extract the time series and do the whole-brain FC analysis. This is what my approach aimed to avoid by doing the actual analysis in native space and then only normalize the result, the Zcorr maps for group analysis. Does this make sense?
Apart from that you are correct with your assumptions regarding Freesurfer and TOPUP. However, I am not sure what you mean with regression maps. Would they be part of my FC analysis?
There might be some reasons to keep analyses in subject space; though, I think when people do this it rarely stays in EPI specifically, and more often in the subject anatomical space. It is true that warping has some affects, but note that even just performing motion correction, which is just EPI-EPI alignment, incurs blurring and smearing of data to start with. In the present case, I have difficulty seeing why analysis in EPI space would be preferred for two reasons:
Firstly, it sounds like your seeds here came from a different space and were warped in already; did that come from a standard space itself?
Secondly, if you plan do to your main analysis in standard space anyways, then it seems like any downsides to warping are only magnified by splitting the process up (that is, incurring extra blurring).
Re. regression maps: a primary output of single subject analysis is the time series from regression modeling (GLMs at each voxel). For task FMRI, this leads to having beta weights+stats at each voxel. For resting and naturalistic FMRI, this leads to having processed time series at every voxel---namely, the residuals of the model that have hopefully had as much non-neuronal influence reduced as possible---which are generally the residuals of the model (the errts time series, from afni_proc.py). That set of time series is what you would use for correlation analysis, in whatever space you are in.
Re. using a high res map to average the EPI: what size are your EPI voxels? Most FMRI voxels are 3 mm isotropic, or perhaps 2 mm isotropic at the smallest. An 0.4 mm isotropic voxel has just 0.8% of the volume of a 2mm isotropic voxel. That is, it will take 125 high-res voxels just to fill a single 2mm isotropic EPI voxel, or 422 high res to fill a 3mm isotropic voxel. Is your ROI the whole brainstem (so, large) or very fine?
I see where you are coming from and I follow your line of reasoning. I was just wondering whether it is preferable to apply a binarized seed (mask), which indeed has been warped from the 0.4 mm highres space via the anatomical image (0.7 mm) to EPI space (1.5 mm) using 3dNwarpApply, to do the FC connectivity analysis (while the EPI time series is still in native space) und get a mainly spared Zcorr map of seed-to-whole-brain correlation coefficients that next can be spatially normalized.
To quickly answer your questions:
Yes, the seeds come from a different space, but highres and not standard space
That is correct overall - for me it was mainly interesting to consider whether it makes a difference if I normalize the whole 4D time series before doing the FC analysis or whether I do the analysis in native space and take only the resulting correlation coefficient map - or regression map (thanks for the explanation!) - which essentially summarizes the relationship between the time series of the seed and rest of brain
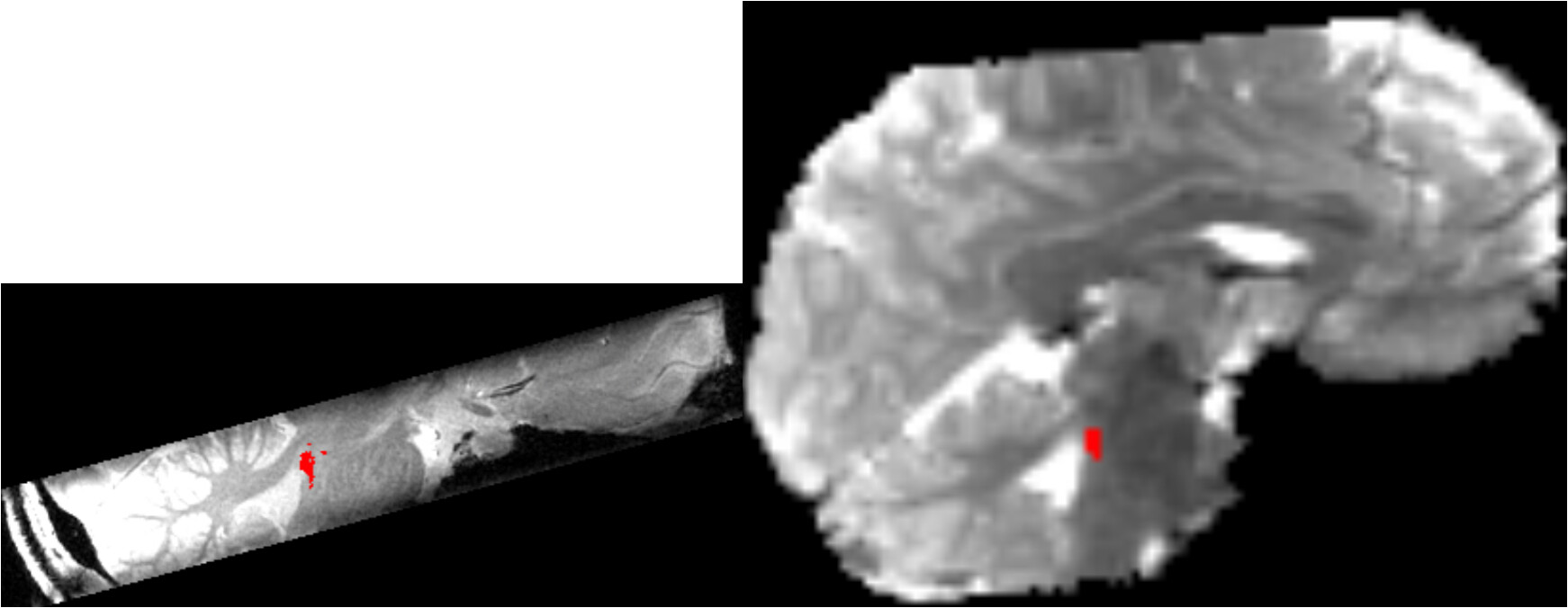
Finally, the seed mask only covers a small area of the brain stem, which looks essentially like this:
where the left image is the 0.4 mm highres space and the right side, the native EPI space. The red spot is the seed mask, which is currently used for the 1st level FC analysis.
Thanks for adding those details. Your final analysis of interest will be in standard space, so I think that it would still be better to interpolate only once. Having a finely structured ROI (note that (1.5/0.4)**3 is still >50) makes it all the more beneficial to only apply a warp once.
Additionally, I think your EPI was acquired obliquely, from the look of that image. I wonder if you have applied that obliquity to it already, before processing? I would not do that, for the same reason cited for much of the recommended processing choices here: that is a regridding process, and therefore inherently blurs the data values. You can put the original EPI into afni_proc.py, and it won't be deobliqued separately, but instead that will be dealt with in a way to not add extra blur.
The
National Institute of Mental Health (NIMH) is part of the National Institutes of
Health (NIH), a component of the U.S. Department of Health and Human
Services.