AFNI version info (afni -ver): AFNI_25.0.11 'Severus Alexander'
I'm using AFNI for the first time for a group project (assessing false positive rates in fMRI analysis via a comparison of 3dttest++ and -etac analyses on pseudostimulus timings/false tasks), and my group and I severely underestimated just how large the learning curve is for AFNI. We've spent about 20 hours debugging our pre-processing code, and the data still isn't in a place where it's usable. I'm having two main issues, one to do with alignment (-cmass) and masking (-mask_epi_anat) - the main questions are below, following my afni_proc.py pipeline.
Data used: Beijing dataset from the 1000 Functional Connectome Project (resting state data).
Here's the overall code we've created (we tried -giant_move to help with our alignment issues):
afni_proc.py \
-subj_id sub01018 \
-copy_anat sub01018/anat/mprage_skullstripped.nii.gz \
-anat_has_skull no \
-dsets sub01018/func/rest.nii.gz \
-blocks tshift align tlrc volreg mask blur scale \
-tshift_opts_ts -tpattern alt+z \
-align_opts_aea -cost lpa -giant_move -check_flip \
-tlrc_base MNI152_2009_template+tlrc \
-volreg_align_to first \
-volreg_align_e2a \
-volreg_tlrc_warp \
-mask_epi_anat yes \
-blur_size 4.0 \
-html_review_style pythonic
#Execute proc.sub01018 script
tcsh -xef proc.sub01018 2>&1 | tee output.proc.sub01018
The above code produces the following (apologies about the screenshot):
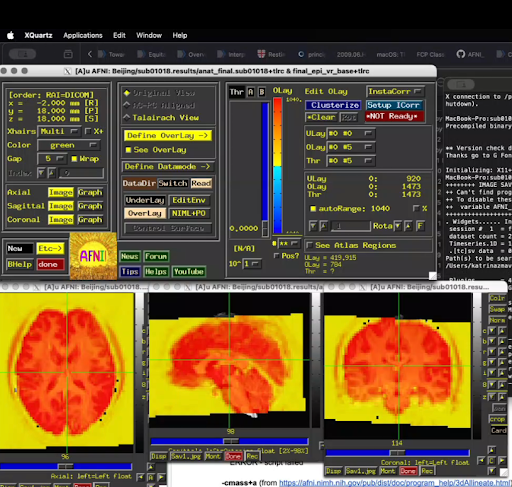
My two main questions:
- Alignment:
I keep getting the warning/message to use -cmass when I run the proc.sub01018 script. However, when I tried adding -cmass to the -align_opts_aea line, I get a script failure error because it's expecting a parameter for -cmass. I then used -cmass+a (as per the 3dAllineate info page, AFNI program: 3dAllineate) and -cmass a, both of which result in a script failure error because it doesn't recognize the "a" after the -cmass command. What's the expected parameter for -cmass? Is it just "yes," similar to the parameter for -mask_epi_anat?
Note: -giant_move did help alignment a bit, but I'd much rather use centre of mass calculations (this seems like a more flexible option given the next step is to loop the code for the rest of the subjects in the dataset), but it's been frustrating having my script fail whenever I try to use it.
For reference, these are the errors I'm receiving:
-cmass a (-align_opts_aea -cost lpa -cmass a -check_flip)
** ERROR: Unknown and Illegal option '-a' :-( :-( :-(
Here's hoping these excerpts from '3dAllineate -help' enlighten:
'-ashift OR }= Apply the shift parameters (#1-3) after OR'
'-autobox = Expand the -automask function to enclose a rectangular'
'-allcost = Compute ALL available cost functionals and print them'
** 3dAllineate failure
** ERROR - script failed
-cmass+a (-align_opts_aea -cost lpa -cmass+a -check_flip)
** error: option -check_flip follows unknown arg #16 (-cmass+a)
** ERROR - script failed
-cmass (-align_opts_aea -cost lpa -cmass -check_flip)
** error: arg #16 (-cmass) requires 1 params, found 0
** ERROR - script failed
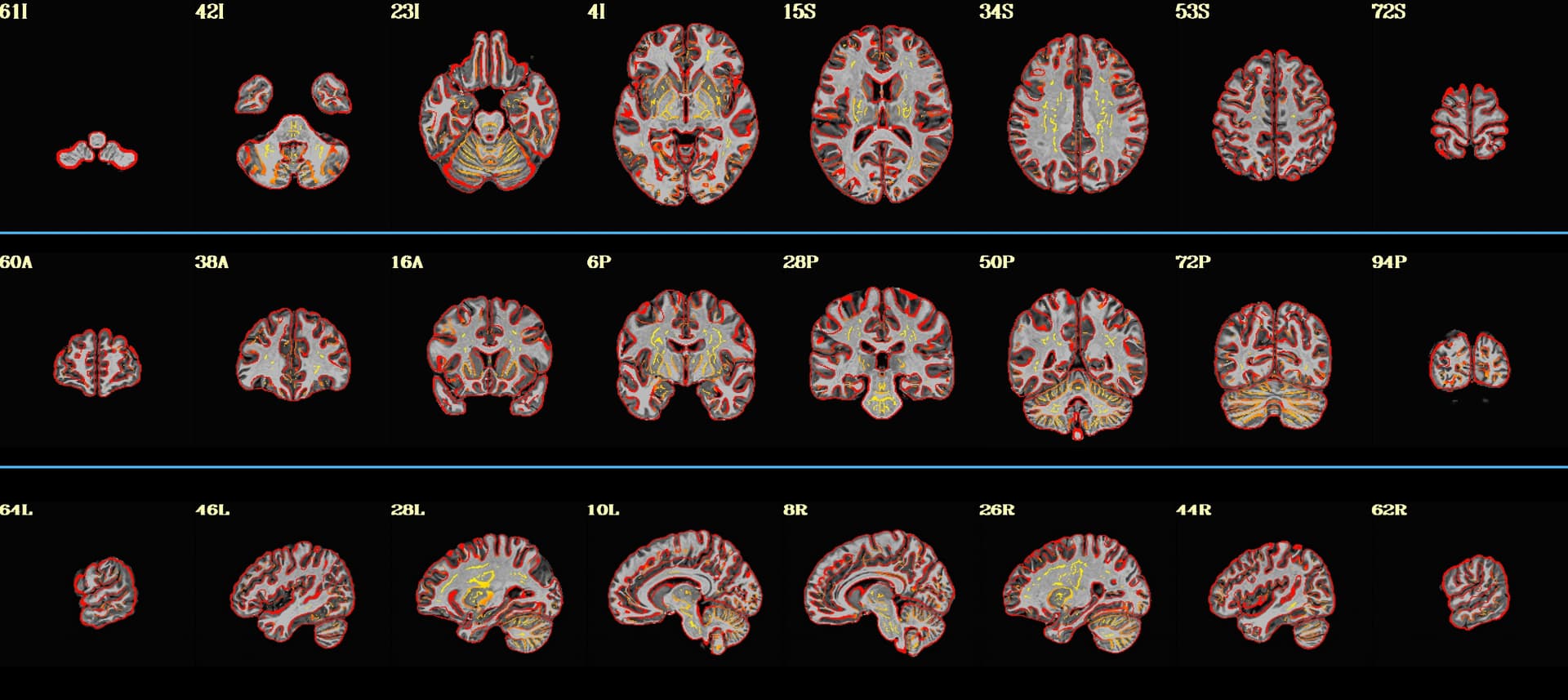
- Masking:
The yellow box surrounding the epi data in non-brain regions didn't go away after I added the -mask_epi_anat yes command (which can be seen in the above image). Do I need to add -mask_apply epi in the line following -mask_epi_anat for the mask to work properly? Or is the excess yellow simply user error and my lack of knowledge on how to finesse the threshold overlay values in the AFNI GUI?
Any guidance would be much appreciated!!